Need help with healthcare project?
Today, the European government (to be more precise, the European surveillance system) has a well-planned strategy that includes both local and individual national requirements. Issues with divergent interpretation of the existing regulations have exposed weaknesses in the current legal system and have reduced patient and healthcare professional confidence in the real safety of devices used by medical personnel.
The new medical regulations are paving the way for a much more people-friendly environment. Patients benefit from innovative, effective devices and even new drugs. The new medical rules introduce better protection for patients and their personal safety. In particular, new devices with a high level of risk are subject to more stringent checks prior to being introduced to the global market.

The rules haven’t changed 180 degrees. Though, the developers of medical software are forced to think of an implementation model in order to comply with the recent change of legislation.
Another thing to keep in mind is the device classification that has also changed a bit. To keep up with the requirements, one will have to review every medical device just to make sure it is classified correctly.
Table of Contents
MDD for Software Development – More Info
The official MDD (Medical Devices Directive) is aimed at creating modalities for the verification, evaluation and research of medical devices and means before releasing them to the market. The purpose of the mentioned document is to minimize risks of injury, sickness, infection.
The document, implemented in the 90’s, establishes compliance assessment procedures according to the classification of products (medical devices), and also establishes mechanisms for authorities to maintain and monitor public health. Under “product” we mean any device, any instrument and substance used by medical personnel.
The official MDD (Medical Devices Directive), which is now replaced by the MDR document, established the developers’ responsibility and the conformity requirements before placing medical products on the EU market for products with the purpose of:
- Diagnostication, checkup, medication, alleviation or prevention of disease
- Diagnostication, checkup, medication, alleviation of handicap or compensation for handicap
- Investigation, replacement or modification of the anatomy or of a physiological process
- Control of ovum fertilization
MDR for Software Development
The official MDR (Medical Devices Regulation) is aimed at ensuring smooth functioning of the market of medical means. The purpose of the mentioned document is the high-level protection of patients. The regulation is setting quality and safety standards for various medical devices and harmonizing the rules for placing and putting such means into service on the EU market. This enables high quality patient care in both public and private clinics.
The document introduces new classification standards, using which the manufacturers have to determine the risk class of their medical products. However, manufacturers should be aware that the risk class may differ from the class assigned under MDD.
The MDR vs MDD – What a Software Developer Should Know
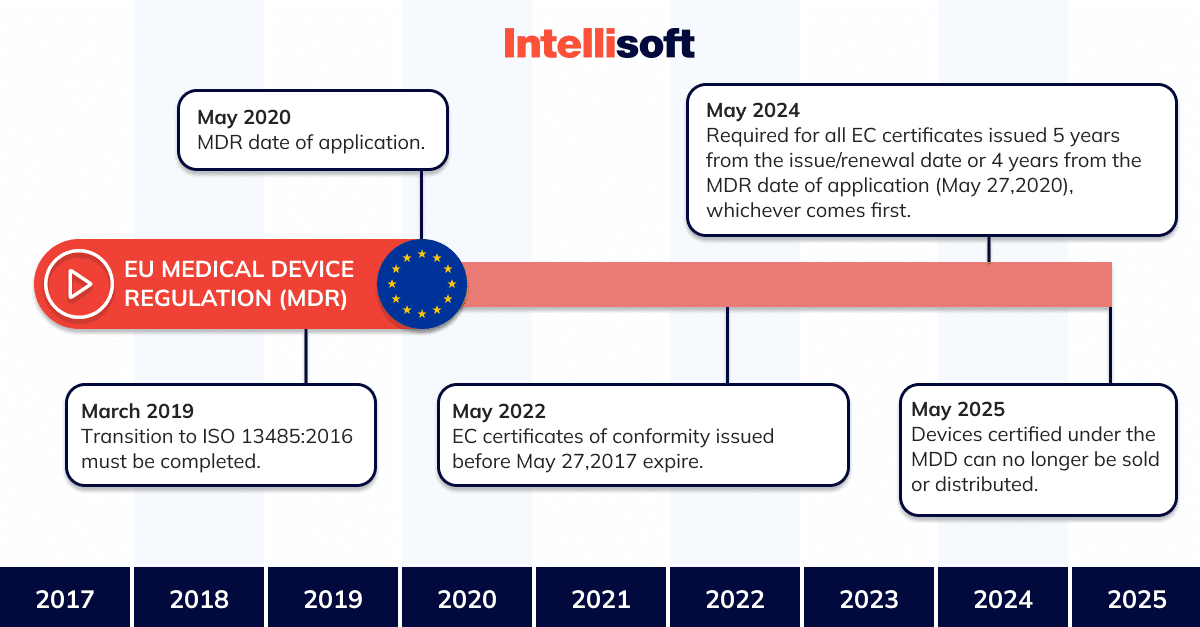
The Medical Device Regulation (or MDR) came into force in May 2017. It replaces the Medical Devices Directive (or MDD), which certifies the supply system quality.
With regard to the practical impact on manufacturers and products, both MDR and MDD generally define the same regulatory requirements. None of the requirements has been completely removed, but some new requirements have been added to the MDR. Compared to the MDD, the MDR places greater emphasis on an approach that takes into account the entire life cycle of a medical device in terms of safety, confirmed by clinical data. The MDR, which is replacing the older document, is establishing stricter requirements for the designation of notified bodies and increasing the control and monitoring by national competent authorities.

In particular, the MDR contains the following points:
- Tighter requirements for bringing medical devices and means to market
- Tighter control on market
- Tighter requirements for notified bodies (for example, DEKRA)
- Manufacturer’s responsibility for the quality traceability, and safety of the medical devices already on the market
- Manufacturer’s responsibility for the fulfillment of warranty requirements, as well as for fixing complaints
- Introduction of UDI marking for unambiguous identification of medical products
- More protection for patients participating in clinical trials
Related readings:
- Healthcare App Developers for Hire: Which Option to Choose
- Best Examples Of Successful Healthcare IT Start-Ups From Denmark
- Catch a Guide on Quality Management System (QMS) for Medical Device Startups
- What Technologies Are Driving the Health Protection System?
- The Internet of Things (IoT) in Healthcare & Medicine
MDD Regulations Now Replaced: Why Was the MDR Introduced by the Government?
As we have mentioned, the European surveillance system has a well-planned strategy. And if issues arise, they know how to solve them. Problems with divergent interpretation of the existing regulations have exposed weaknesses in the current legal system. These problems reduced both patient and personnel confidence in the actual safety of the medical devices being used.
To address this issue, in 2012 the Commission has proposed some changes on products that are used for medical purposes. The provisions mean much more control to ensure high level patient care, at the same time promoting innovation and increasing the competitiveness of the technology sector.
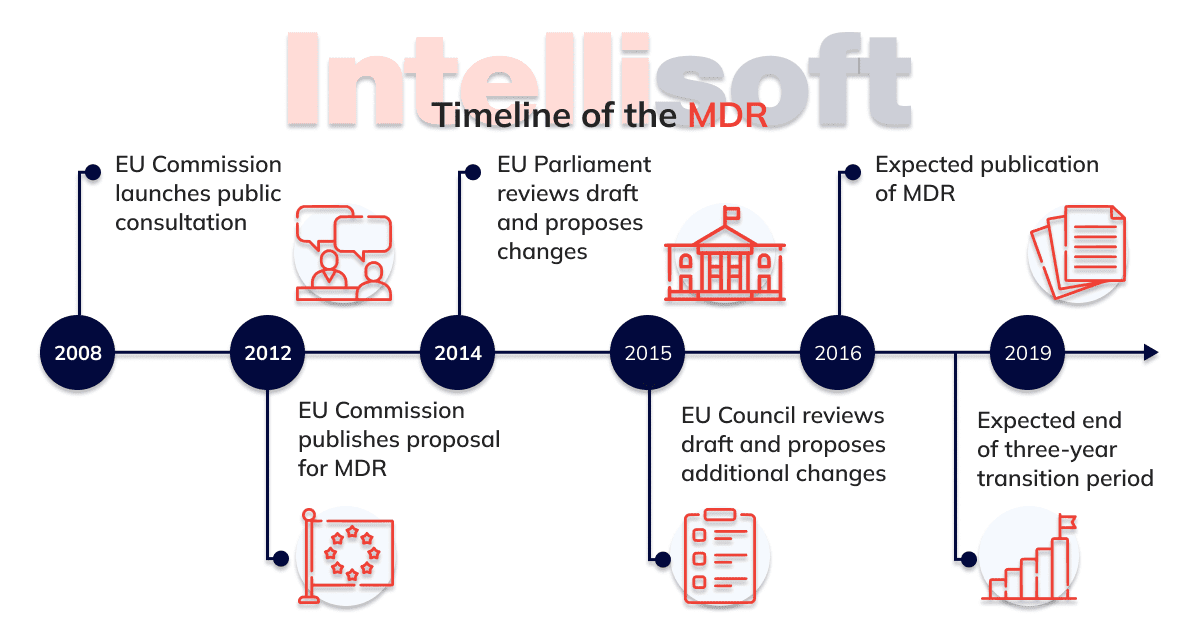
Being tightened are rules on clinical evaluation and clinical trials (as well as for in vitro efficiency studies). Being strengthened and stringent are also the requirements for the use of dangerous substances.
A comprehensive database on medical devices (the EUDAMED) has to provide the real picture of the life cycle of all products available on the EU market. Thanks to the current EU system, absolutely all medical devices and the necessary accessories are subject to the new rules.
The MDR versus MDD: Implementation of New Requirements
- First, determine the scope and class of the medical device that will be developed (remember being careful about these decisions, because they will influence all the subsequent actions).
- Secondly, determine the stages of the software development and all the resources necessary for the successful project implementation.
- Risk minimization, as well as verification of compliance with general security and performance requirements. This step should begin with risk management planning, ending with a market surveillance plan.
- Clinical assessment. Further actions depend on the class and novelty of the medical device in plan, but a post-market clinical follow-up plan is mandatory.
- Drawing up technical documentation. Most of the documentation will be generated in steps 1 to 4, so do not worry.
- Drawing up agreements on the distribution of products. This stage implies the fulfillment of the rules of other product manufacturers, the obligation to exchange information with them and the appointment of a representative in the government.
- Registration of the product and the manufacturer firm with the competent authorities. You will get a registration number, the UDI data uploaded to the official database, and a declaration of conformity of the EU.
- Verification of necessary documentation and start of export.
Fulfillment of the obligations in accordance with the new MDR involves the following points:
- The support of the so-called risk management concept
- The fulfillment of post-sale clinical activities
- The update of all technical documentation
- The improvement of the product’s quality
Why Is It Important to Choose a Suitable IT Team for Your Healthcare Project
As you can see, there are many factors to consider when designing medical devices or means. According to the experience of many software companies, it gets more complicated with a wrong assembled IT team. Therefore, we strongly recommend you to take the necessary time on finding suitable specialists and organizing subsequent interviews. But if you don’t want to waste time and energy on headhunting, consider other popular alternatives.
How about simply transferring your project to experts? Outsourcing certain business processes might be the solution. By contacting real experts with the necessary knowledge and experience, you get the chance to focus on your business goals. This might not only save time and money, but make your company more productive.
Choose your outsourcing partner considering the following factors:
- Field expertise. Don’t choose a company with experience in transportation for the development of medical devices.
- Customer reviews. Take time to read customer reviews.
- Realistic scope and deadlines. Don’t trust those providers who are not able to calculate the scope and deadlines of a tech project.
- Project budget. Outsourcing development is all about efficacy, time and cost savings.
How We Can Help
Does your company provide medical services? Do you get the feeling sometimes that it is time to introduce advanced technology to provide medical services of a higher level? You’re lucky. Our specialists have expertise in developing software products for the medical sector.
Don’t wait to bring your ideas to life. Contact us for a free consultation!