In May 2021, the EU Medical Device Regulation (MDR) replaced previous MDD and AIMDD directives. This pivotal regulation represents a significant overhaul of the regulatory framework governing the development, manufacture, and distribution of medical tools within the EU.
Our blog article will cover the crucial components of EU regulatory compliance, its objectives, the significant changes it brings to the MedTech industry, and its challenges and opportunities. IntelliSoft has some solid healthcare cases, like the one delivered to our client, Daintel. For these clients, we also ensured MDR compliance.
Table of Contents
EU MDR – What Is It and Why Is It Necessary?
For the first time in over two decades, the European Union has enacted a major overhaul of its regulatory framework for medical devices, launching the Medical Device Regulation EU No. 2017/745 alongside the In Vitro Diagnostic Regulation (IVDR). These new regulations, which were implimented on May 26, 2021, for the EU MDR and May 26, 2022, for the IVDR, supersede the previous Medical Device Directives (MDD) and are uniformly enforced across all 27 EU member states.
New directives have been established for selling medical tools in the EEA. All so-called ‘legal manufacturers’ must follow strict safety and performance criteria to ensure their products meet the highest standards. The shift to the EU MDR was propelled by the urgent need for more contemporary and rigorous medical device regulations within the EU, prompted by several significant factors:
- Technological Advancements. The rapid pace of innovation in medical technology has introduced complex devices with new functionalities and applications. Traditional regulatory frameworks designed for earlier generations of medical devices struggled to adequately address the safety, efficacy, and quality assurance of these advanced products. The EU MDR was developed to ensure that regulatory oversight keeps pace with technological progress, incorporating provisions for software as a medical device and other innovative technologies.
- Global Harmonization. There has been a growing global movement towards harmonizing medical device regulations to facilitate international trade and ensure consistent safety standards worldwide. The EU MDR aligns more closely with international guidelines, such as those proposed by the International Medical Device Regulators Forum (IMDRF), helping streamline regulatory processes for manufacturers operating in multiple jurisdictions and ensuring high protection for EU citizens.
- Safety Incidents and Public Health Concerns. High-profile safety incidents involving medical devices, such as the PIP breast implant scandal and issues with metal-on-metal hip replacements, highlighted vulnerabilities in the existing regulatory framework. These events heightened public and regulators’ attention, highlighting the necessity for stronger pre-market assessments and ongoing post-market monitoring to more efficiently identify, evaluate, and respond to safety concerns.
- Deficiency of ISO 13485. ISO 13485 sets forth the standards for quality management in medical tools. Furthermore, it needs to encompass the full scope of oversight needed to assess the safety and effectiveness of these devices over their entire lifecycle. The EU MDR fills this gap by implementing regulations for medical devices that evaluate clinical evidence, post-market surveillance, risk management, and enhanced transparency. These additions aim to safeguard patient safety and ensure the effectiveness of products influenced by the latest technological improvements
- Need for Increased Transparency and Traceability. Before the implementation of the EU MDR, the medical device market suffered from a noticeable opacity, hindering patients, healthcare professionals, and regulatory bodies from obtaining information about medical devices. The EU MDR tackles this problem by establishing more apparent transparency measures by launching the European Database on Medical Devices (EUDAMED) to simplify the retrieval of tool information. Moreover, the Unique Device Identification (UDI) system is introduced to improve gadgets’ traceability over their entire lifecycle.
The EU Medical Device Regulation is a comprehensive guide for medical device regulation that aims to ensure the safety and effectiveness of products. It includes 10 chapters, 123 articles, and 17 annexes that manufacturers must meet. The regulation provides a precise framework, guiding manufacturers through the complex landscape of medical device compliance.
What Are the Major Thematic Changes in the EU Medical Device Regulation?
European regulations for medical devices represent a transformative change from the previous Medical Device Directive (MDD). Rather than concentrating primarily on pre-market approval, the MDR adopts a comprehensive lifecycle approach to regulating medical gadgets.
The Medical Device Directive (MDD) was a guidebook for manufacturers that outlined the necessary steps to obtain CE marking, which was required to enter the European Union (EU) market. However, the new EU medical device regulation has expanded the scope of accountability. It now enforces a more stringent pre-market evaluation process and emphasizes continuous post-market surveillance. This approach is designed to ensure that companies remain vigilant about the safety and effectiveness of their products at every stage, from the initial conception to clinical use and beyond.
The European market, consisting of 27 member states — exclusive of the United Kingdom — along with associated members within the European Economic Area (EEA), such as Liechtenstein, Norway, and Iceland, is home to a population exceeding 500 million, many of whom are part of an aging demographic. As the population ages, the susceptibility to potential risks posed by medical devices increases, making the comprehensive oversight of the MDR not just prudent, but essential.
The escalating healthcare demands of this population, and the advancing intricacies of medical technologies, highlight the significance of the MDR’s emphasis on the entire product lifecycle. This proactive strategy aims to protect patient health across all stages of medical tool usage, affirming the EU’s dedication to keeping up-to-date patient safety standards amidst demographic changes and technological progress.
H3 How the MDD Differs From the MDR
The Medical Device Directive (MDD), and European regulations for medical devices, serve as foundational regulatory frameworks within the EU. While they share common objectives, the MDR has been established as a successor and an enhancement of the MDD. The MDR builds upon the MDD’s provisions, incorporating more extensive classification rules and broadening the spectrum of device classes.
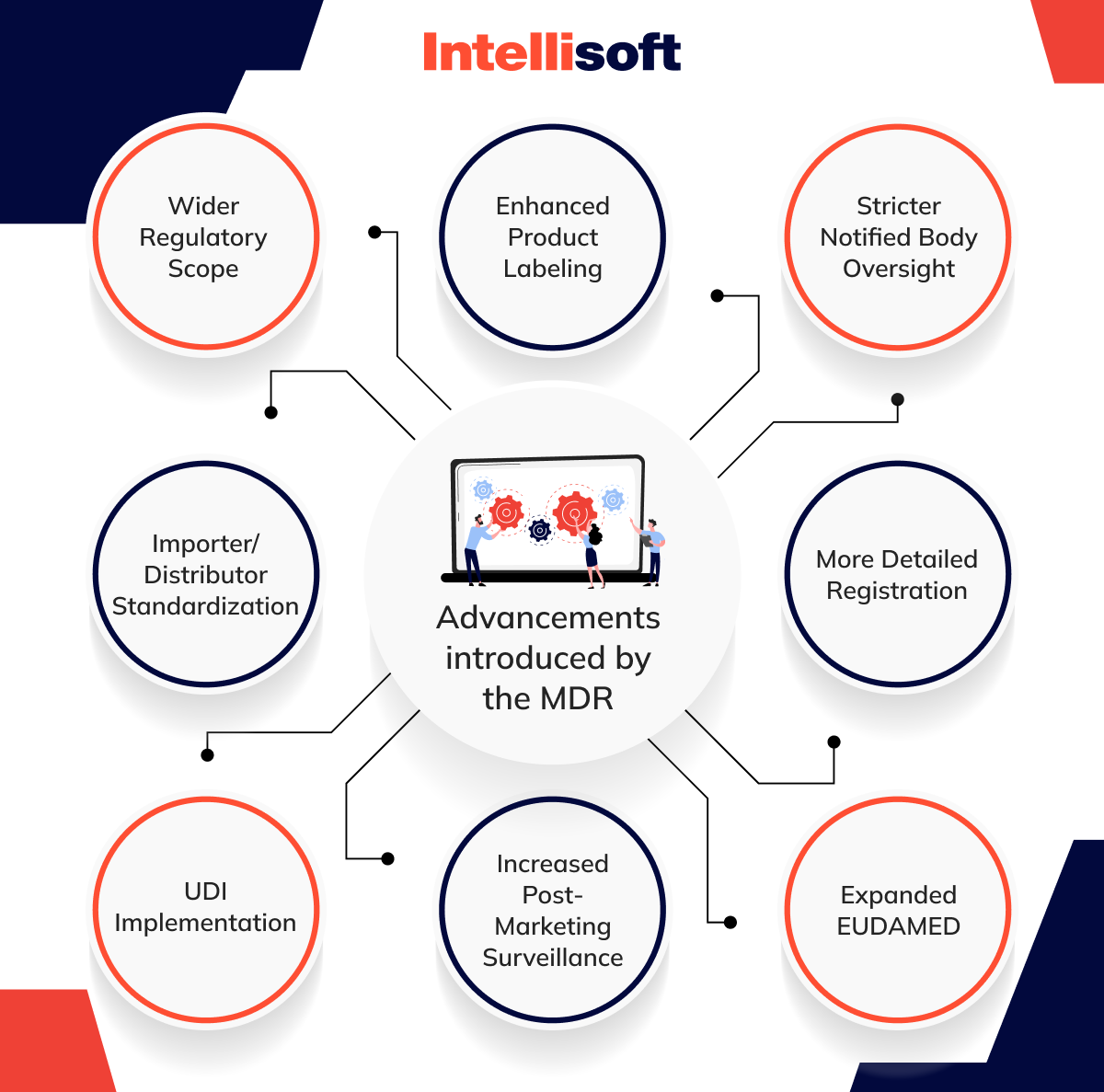
Other crucial advancements introduced by the MDR cover:
- Enhanced Product Labeling. The MDR mandates clearer labeling to inform users and healthcare professionals better.
- Stricter Notified Body Oversight. Criteria for notified bodies are more rigorous, improving assessment reliability.
- More Detailed Registration. Registration under the MDR demands more comprehensive safety and performance data.
- Expanded EUDAMED. EUDAMED now serves as an extensive, public database for medical gadget data.
- Increased Post-Marketing Surveillance. The EU MDR medical device regulation requires frequent PMS reporting based on device classification.
- UDI Implementation. Unique Device Identification is now required for improved tool traceability.
- Importer/Distributor Standardization. The MDR sets clear guidelines for EU importers and distributors.
- Wider Regulatory Scope. The new regulation has expanded its scope to include products not previously considered medical tools under its classification.
Overview of the EU Medical Devices Regulation (MDR) implementation
The Medical Device Regulation, EU is an essential update to the framework that governs the approval, oversight, and monitoring of medical tools in the European Union. It was activated in May 2021 and replaces previous directives with a more stringent and transparent system that enhances patient and user protection.
The MDR is significant in the healthcare industry as it improves patient safety by implementing strict criteria for pre-market assessments and ongoing post-market surveillance. It emphasizes the importance of clinical proof for the approval of medical gadgets, ensuring that only rigorously tested and safe products are introduced to the market. This approach raises patient safety standards and builds trust in the medical technology field.
Moreover, the MDR stresses the industry’s importance of transparency and traceability. It establishes a comprehensive EUDAMED database implemented across the EU to achieve this. This database streamlines the monitoring process and provides public access to detailed information about all products available in the EU market. The MDR aims to improve product safety and efficacy and enhance public confidence in medical technology.
The enforcement of the MDR has significant implications for the global medical technology (MedTech) industry, given the size and impact of the European market. The regulation affects European manufacturers and international entities aiming to enter or maintain operations within Europe. EU regulatory compliance with the MDR is crucial for MedTech companies seeking to tap into or maintain their market share in this highly competitive space.
Transitional Provisions
Certain medical tools and IVDs may still be sold under the old Directives, referred to as “legacy devices.” The timeframes for such sales have been extended for both Regulations. The IVDR’s extension was arranged in January 2022, while the MDR’s extension happened in March 2023. These gadgets now have more time to conform to the new regulations and obtain a CE marking.
The recently introduced extension in the MDR has relieved manufacturers and patients. The extension provides a more extended timeframe for notified bodies to certify medical tools and allows their entry into the market. This development is significant since meeting the regulatory requirements within the specified deadlines has proven challenging for manufacturers. The previous deadlines posed a risk to patient safety as it could have led to the withdrawal of many products from the market. However, the MDR’s transitional period extension applies only to safe products.
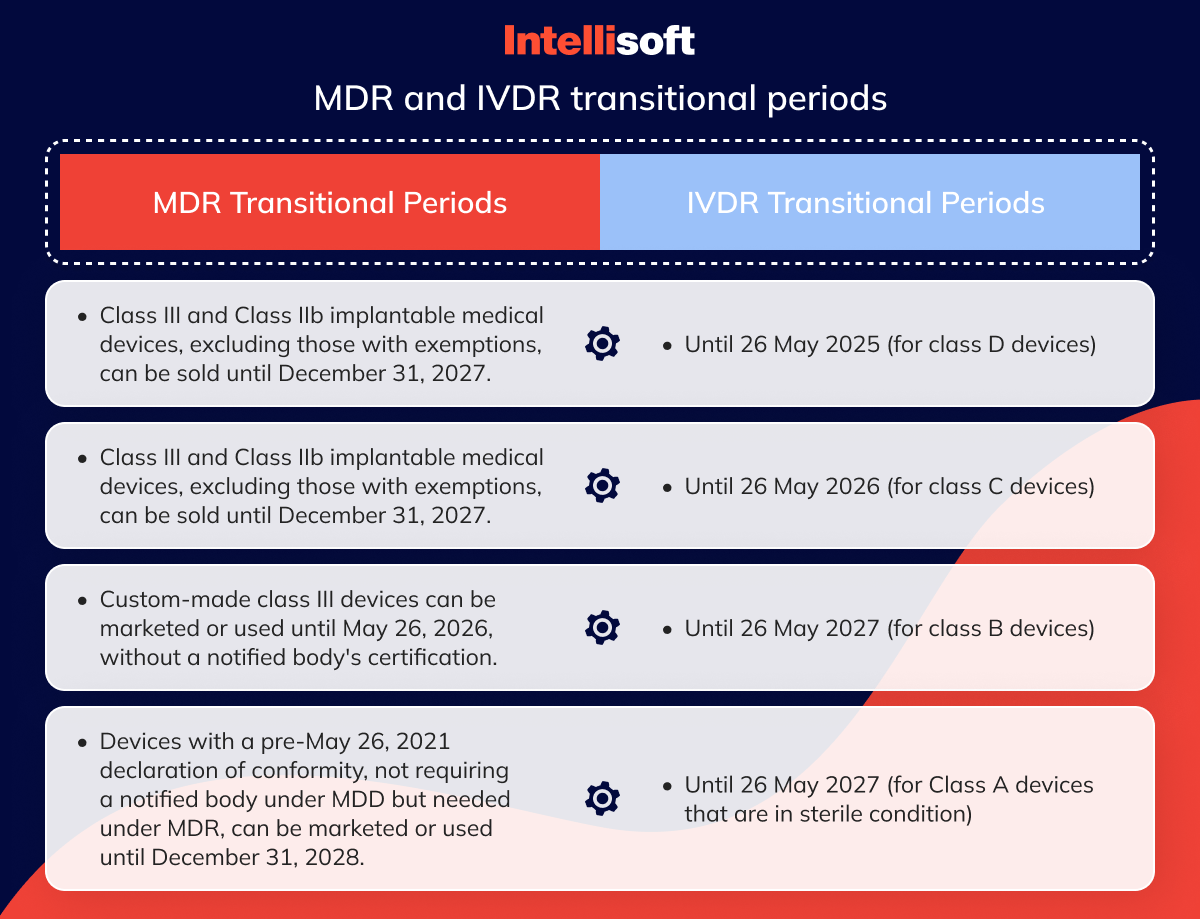
MDR transitional periods
The length of the extension period is determined by the risk category assigned to the device
Manufacturers can sell Class III medical and Class IIb implantable devices until December 31, 2027. Nonetheless, certain tools are exempt from the EU MDR medical device regulation due to their foundation in well-established technologies.
Manufacturers can sell products from other risk classes until December 31, 2028.
Custom class III medical devices can be placed on the market or put into service until May 26, 2026, without a certificate from a notified body.
Manufacturers may place on the market or put into service devices with a declaration of conformity drawn up before May 26, 2021, which did not require a notified body’s involvement under the MDD but do under the MDR until December 31, 2028.
These transition periods are only allowed for specific tools, as outlined in Regulation (EU) 2023/607.
IVDR transitional periods
The duration of the extension granted will vary depending on the risk classification of the in vitro diagnostic product.
- Until 26 May 2025 (for class D devices)
- Until 26 May 2026 (for class C devices)
- Until 26 May 2027 (for class B devices)
- Until 26 May 2027 (for Class A devices that are in sterile condition)
Key Differences and Similarities Between EU MDR and US FDA Regulations
The EU and the United States operate under unique and complex regulatory frameworks to oversee medical products. Both systems have similar objectives under EU MDR and US FDA regulations and are meticulously designed to guarantee the safety and effectiveness of medical tools, a cornerstone in their shared mission to protect public health. This commitment implies strict monitoring of medical tools, ensuring their safety before reaching consumers and healthcare professionals.
Similarities:
- Quality System Requirements. It is worth noting that the Food and Drug Administration (FDA) and the European Union Medical Device Regulation (EU MDR) have underlined the crucial role of quality systems for medical products that conform to ISO standards. This emphasis is critical as it ensures that such products are meticulously designed, manufactured, and distributed in strict compliance with stringent safety and quality standards. In other words, adhering to ISO standards guarantees that medical products are thoroughly scrutinized, tested, and verified to meet the highest safety and quality criteria, safeguarding patients’ health and well-being.
- Risk Requirements. Both regulatory frameworks prioritize assessing and mitigating potential risks associated with medical equipment to safeguard patient well-being through robust safety measures across the medical tool supply chain.
Differences:
- Regulatory Pathways. The Medical Device Regulation EU classifies products into four categories based on risk, requiring a conformity assessment that may involve a Notified Body for most tool classes. This system emphasizes a rigorous evaluation based on the device’s potential risk to patients. In contrast, the FDA categorizes gadgets into three classes and utilizes specific pathways for device approval—Premarket Approval (PMA) for high-risk products and 510(k) clearance for lower-risk devices, focusing on demonstrating substantial equivalence to previously approved tools.
- Centralized vs. Decentralized Approval. The approval process under the EU MDR is characterized by its decentralized nature, with Notified Bodies in individual EU member states assessing tool conformity. This contrasts with the FDA’s centralized approval mechanism, where the agency is the sole authority for approving all medical devices marketed in the U.S., ensuring a uniform evaluation standard nationwide.
- Clinical Data Requirements. EU MDR mandates extensive clinical evidence to support the safety and performance of tools, requiring continuous clinical evaluation and, if necessary, clinical investigations, particularly for higher-risk devices. This is part of a broader emphasis on post-market surveillance. The FDA, while also requiring clinical data to demonstrate safety and effectiveness, particularly for PMA applications, allows 510(k) clearances based on substantial equivalence to existing gadgets, which may not require new clinical trials if adequate predicate device data exists.
Related articles:
- Understanding the Differences Between MDD vs MDR: The European Regulation
- Healthcare App Development Guide: Transforming Patient Care with User-Centric Solutions
- Automation in Healthcare
- A Step-by-Step Guide to Developing HIPAA-compliant Medical Apps
- Inside Out: Exploring the World of Diagnostic Medical Imaging Software
MDR Challenges and Implications for Medtech Firms
The Medical Device Regulation (MDR) introduced in the EU in May 2021 has posed several challenges for the US and other international medtech firms, affecting their market access and strategic approaches significantly.
Challenges introduced by the Medical Device Regulation:
- Increased Complexity for CE Mark Approval. The new EU medical device regulation has made obtaining a CE mark to market medical tools in the European Union (EU) more complex and demanding. This change has increased the regulatory burden and made the EU market less attractive to manufacturers.
- Stringent Clinical Data Requirements. One of the significant changes is the requirement for more rigorous clinical data to support the safety and efficacy of medical gadgets. This shift in the regulatory landscape means that companies must invest more in clinical trials and data collection to comply with the MDR.
Impact on Market Access and Company Strategies:
Shift Towards FDA Authorization. Medtech companies find it increasingly challenging to abide by the Medical Device Regulation (MDR). As a result, there has been a noticeable shift in focus towards obtaining authorization from the US Food and Drug Administration (FDA). The US market’s streamlined approval processes and perceived straightforward regulatory environment make it more attractive to MedTech companies.
Strategic Re-evaluation. Companies are adapting strategies to comply with new medical device regulations. This means reallocating resources to less regulated markets or investing in R&D to meet MDR demand.
How To Determine if Your Software Qualifies as a Medical Device Under the EU MDR?
The EU MDR states that software intended for one or more medical purposes can be classified as a medical tool. However, even if it is used in a healthcare setting or for lifestyle and well-being purposes, software for general purposes is not considered a medical device. The primary factor in determining whether the software is a medical device is its Intended Use.
As a side note, it is essential to mention that the Intended Use is a document that specifies the predetermined usage of a particular medical product. It is essential to prepare a certification strategy and assess the class of the product accordingly.
Based on your software’s Intended Use, you can now determine whether your software is a medical device following particular guidelines.
The software can be classified as a medical device if it meets one of the following criteria:
- It is explicitly identified in the EU regulations for medical devices.
- It controls or influences a medical device.
- It assists post-processing or data preparation, such as analyzing ECG data.
- It calculates output signals or values related to diagnostic or therapeutic purposes.
- It supports objective diagnosis or treatment.
On the other hand, software is not considered a medical device if:
- It is used for administrative purposes, such as managing patient data.
- It is used for training medical professionals with medical knowledge.
- It is used for general maintenance of medical gadgets or their components, although it may sometimes be treated as medical device accessories.
- It is used as a development or production tool, although such tools must get validation.
- It represents a proprietary operating system.

How to Classify a Medical Device Under the EU MDR?
The EU Medical Device Regulation (MDR) of the European Union establishes a comprehensive framework for the categorization of medical devices based on the degree of risk they pose to patients and users. To classify a medical tool according to the EU MDR, one must familiarize oneself with the regulation’s three primary classes: Class I, Class II (further divided into Class IIa and IIb), and Class III. Each class corresponds to a different level of risk.
The first step in classifying a medical device is ensuring we have all the necessary documents and information. The primary documents that need to be prepared and collected before initiating the classification process include:
- Device description, including its Intended Use
- Preferred usage and user group
- Data on patients and diseases affected by the tool
- The treatment
- Diagnostics and monitoring
Based on this information, you can assess your device’s class.
Within the framework of the EU MDR, the classification of a medical device’s risk level is governed by a structured rules-based system, detailed in Annex VIII of the MDR. This system encompasses 22 rules, systematically organized into four distinct sections, each dedicated to a specific category of gadgets, ensuring precise risk assessment based on device type and usage.
Rules 1-4: Non-Invasive Devices
These tools do not breach the body’s surface or enter any orifice for their function.
Rules 5-8: Invasive Devices
This category includes tools that either partially or fully penetrate the body through any surface or orifice.
Rules 9-13: Active Devices
Devices falling under this section depend on an external energy source other than what the human body can provide for its operation.
Rules 14-22: Special Category
This unique section accommodates tools that do not conform to the classifications of the first three categories, covering a broad spectrum of specialized medical tools.
Furthermore, the MDR specifies three durations of intended use for tools, which play a crucial role in determining the applicable classification rules:
- Transient. Products designed for continuous operation for less than 60 minutes.
- Short-term. Products intended for continuous use spanning from 60 minutes up to 30 days.
- Long-term. Products meant for constant use extending beyond 30 days.
This comprehensive and categorically segmented system under the EU MDR ensures that every medical device is accurately classified according to its risk level, considering its operational nature and duration of use. This meticulous approach enhances regulatory oversight, ensuring the safety and efficacy of medical devices within the EU.
Class I Devices
Class I products have the lowest risk associated with their use. These are typically non-invasive tools and must adhere to general controls to ensure safety and efficacy. An example of a Class I device would be a non-sterile, manual examination instrument.
Class II Devices
Class II products are subdivided into Class IIa and Class IIb, ranging from medium to high risk.
- Class IIa Devices. These medium-risk devices may contact patients directly. They serve a therapeutic, diagnostic, or monitoring purpose and require mandatory compliance assurance with EU medical device regulations. An example of a Class IIa device would be a hearing aid.
- Class IIb Devices. These devices fall under the medium-to-high-risk category. They are similar to Class IIa tools regarding their contact with the patient and purpose but carry a higher risk profile. An example of a Class IIb device might be a blood transfusion bag.
Class III Devices
Class III products are considered to have the highest risk. They are typically invasive, connecting directly with the central circulatory or nervous system, or may be implantable. These devices require a stringent mandatory compliance assurance process due to the potential high risk associated with their use. A typical example of a Class III device is a coronary stent.
Classifying a medical tool under the EU MDR is a critical step in the regulatory process. It determines the level of assessment and compliance activities needed before a product can be marketed in the EU.
How To Ensure Medical Devices Comply With Recent EU Medical Device Regulations?
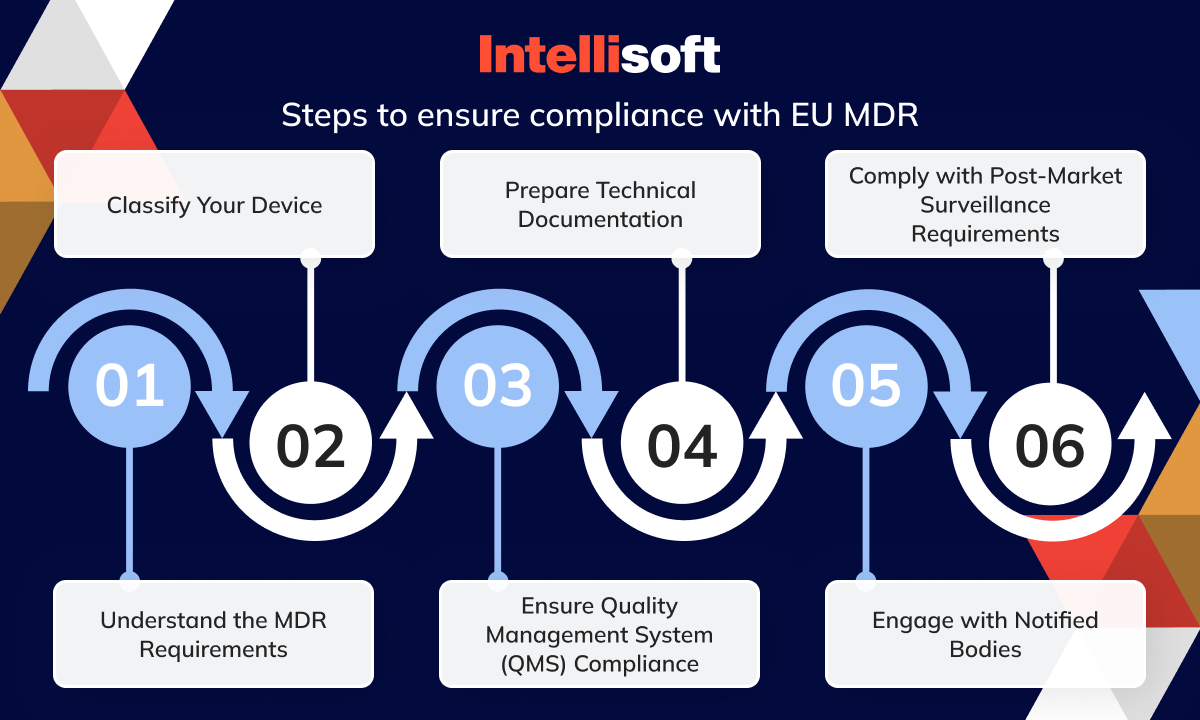
Ensuring compliance with the recent EU Medical Device Regulation (MDR) involves steps and adherence to stringent requirements to improve product quality and safety. Here are the vital steps to ensure EU regulatory compliance:
- Understand the MDR Requirements. Familiarize yourself with the detailed requirements of the MDR, focusing on high standards of quality and safety for medical devices produced in or supplied to Europe.
- Classify Your Device. Determine the class of your medical tool according to the MDR’s risk-based classification system, which may affect the level of scrutiny and regulatory requirements your device will undergo.
- Ensure Quality Management System (QMS) Compliance. Your QMS must comply with the MDR’s requirements, likely aligning with ISO 13485 standards. This system should cover all aspects of your device’s design, production, and post-market surveillance.
- Prepare Technical Documentation. Compile comprehensive technical documentation that demonstrates your device’s conformity with the regulation. This set includes clinical evaluation, risk management, and manufacturing process information.
- Engage with Notified Bodies. You must work with a Notified Body for conformity assessment for specific tool classes. Choose a Notified Body authorized under the MDR to evaluate your device’s compliance.
- Comply with Post-Market Surveillance Requirements. Implement an effective post-market surveillance system to continually assess the safety and performance of your gadget once it is on the market and report any incidents or corrective actions as required by the MDR.
By following these steps and maintaining a proactive approach to compliance, manufacturers can ensure their medical devices meet the MDR’s rigorous standards, safeguarding public health and patient safety.
Conclusion
The implementation of the EU Medical Device Regulation is a significant moment for the entire industry. It introduces more stringent requirements for clinical evidence, transparency, and traceability, which sets a new standard for the safety and effectiveness of medical gadgets available in the European market. The transition presents challenges for manufacturers, but also creates opportunities for innovation and enhances patient safety, ultimately strengthening trust in the medical device sector.
In this changing regulatory landscape, IntelliSoft stands out for its extensive expertise and comprehensive healthcare solutions. We offer tailored software solutions and consulting services that assist companies in achieving and maintaining compliance with the MDR. Our team is equipped with extensive knowledge of the regulation’s intricacies, and we offer a range of outsourcing software development and consulting services designed to simplify the compliance process. From managing documentation to ensuring the traceability of devices, IntelliSoft’s innovative approach simplifies the complex demands of MDR compliance, allowing manufacturers to focus on developing life-saving medical tools.
Our emphasis on data security, continuous tech support, and adherence to regulatory standards positions IntelliSoft as a comprehensive solution provider in the medical device development realm.
Contact us if you want to develop a medical device and need software that complies with European regulation rules.